
EORTC 1209-EnTF (Nintedanib)
A phase II study exploring the safety and efficacy of nintedanib (BIBF1120) as second line therapy for patients with either differentiated or medullary thyroid cancer progressing after first line therapy. EORTC 1209-EnTF.
| Protocolnummer: | EORTC 1209-EnTF |
| Eudractnummer: | 2012-004295-19 |
| METc nummer: | |
| Onderzoekscode: |
Samenvatting
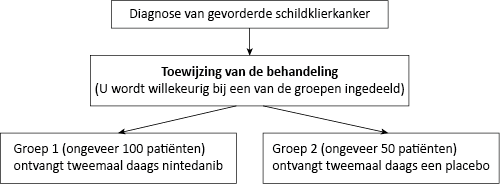
Nintedanib is een geneesmiddel dat de tumorgroei bestrijdt. Er bestaat weinig informatie over het gebruik van nintedanib als een manier om schildklierkanker te bestrijden. Deze studie is bedoeld om te ontdekken of nintedanib helpt om de groei van schildklierkanker te beperken en patiënten met schildklierkanker helpt om langer te leven. In deze studie ontvangt de patiënt ofwel het nieuwe middel, nintedanib, ofwel de placebobehandeling.
Inclusiecriteria
- Door de patholoog histologisch bewezen gedifferentieerde of medulaire schildklierkanker.
- Beschikbaar tumormateriaal ten tijde van diagnose voor histologische review. De voorziening van tumormateriaal voor histologische review is verplicht voor elke locatie.
- Lokaal gevorderde of gemetastaseerde ziekte ongeneeslijk geacht door chirurgie, radiotherapie en/of RAI.
- Geen aanwezige hersenmetastasen, indien eerder aanwezig dan moeten deze tenminste 2 maanden voor randomisatie zijn behandeld. CT of MRI scan van de hersenen is verplicht (binnen 4 weken voorafgaand aan randomisatie) om de mogelijke aanwezigheid van hersenmetastasen vast te stellen.
- Patiënten moeten een meetbare lesie met gedocumenteerde progressie gedurende 12 maanden voorafgaand aan randomisatie volgens RECIST V.1.1.
- Patiënten moeten 1 of 2 (maar niet meer dan 2) eerdere behandellijnen hebben ontvangen en moeten tenminste 4 weken van de behandeling af zijn voorafgaand aan randomisatie.
- Leeftijd ≥18 jaar
- Performance status (PS) 0-1 (WHO)
- Levensverwachting van meer dan 12 weken
- Geen eerdere maligniteiten in de afgelopen 5 jaar, behalve adequaat behandelde carcinomen in situ van de carvix of basaalcel- of spino cellulair carcinoom van de huid
- Geen behandeling gerelateerde toxiciteit ten gevolge van een eerdere behandeling > graad I (behalve alopecia)
- Goede orgaanfunctie, bewezen door de volgende lab uitslagen binnen 3 weken voor randomisatie: (patiënten met een buffer bereik van de normaalwaarden van +/- 5% hematologie en +/- 10% biochemie [met de uitzondering van de glomerulaire filtratie ratio] zijn acceptabel)
- Absoluut aantal neutrofielen > 1500 cellen/mm3
- Aantal bloedplaatjes > 100.000 cellen/mm3
- Hemoglobine > 8.5 g/dL
- Totale bilirubine binnen de normaalwaarden
- SGOT (AST), SGPT (ALT), en alkaline fosfatase ≤ 1.5× ULN (of ≤ 2.5× ULN indien er lever metastasen aanwezig zijn)
- GFR ≥ 45 ml/min volgens Cockcroft en Gault Formule
- Proteinurie CTC-AE < 2
- Coagulatie parameters: Internationale normaalwaarden ≤ 2, PT en PTT ≤ 1.5x institutioneel ULN
- Geen geschiedenis of belangrijke cardiale ziekten zoals:
- Symptomatische CHF (NYHA gradering III-IV)
- Hoog risico op ongecontroleerde ritmestoornissen, d.w.z. atriale tachycardie met een hartslag van > 100/min bij rust, significante ventriculaire aritmie of hooggradig AV-blokkade (tweede graads AV-blokkade Type 2 [Mobitz 2] of derde graads AV-blokkade)
- Geen verlenging van de gecorrigeerde QT interval (QTc) > 480 msec
- Geschiedenis van mycoard infarct 12 maanden voor randomisatie
- Klinisch significante hartklepafwijkingen
- Geen angina pectoris die anti-angina behandeling behoeft
- Geen huidige ongecontroleerde hypertensie (aanhoudend systolie > 180 mmHg en/of diastolie > 100 mmHg) (met of zonder medicatie). Initiatie of aanpassing van bloeddrukverlagende medicatie (s) wordt voorafgaand aan het onderzoek toegestaan.
- Geen bewijs voor actieve bloedingen of bloedingsdiathese
- Geen cerebrovasculair accident op enig moment in het verleden, TIA, diepe veneuze trombose (DVT) of longembolie in de afgelopen 6 maanden
- Therapeutische anti-coagulatie (behalve een laaggedoseerde heparine en/of heparine flush als nodig voor het onderhoud van een inwendig intraveneuze apparaat) of anti-bloedplaatjes therapie (behalve lage dosis behandeling met acetylsalicylzuur <325 mg per dag) is niet toegestaan
- Geen geschiedenis van klinisch significante gastro-intestinale stoornissen inclusief: malabsorptiesyndroom, majeure resectie van de maag of dunne darm die de absorptie van de onderzoeksmedicatie kunnen beïnvloeden, actieve maagzweer, bekende intraluminale metastatische laesies met kans op bloeden, inflammatoire darmziekte, ulceratieve colitis, of andere gastro-intestinale aandoeningen met een verhoogd risico op perforatie. Geen geschiedenis van abdominale fistels, gastro-intestinale perforatie, of intra-abdominaal abces binnen 28 dagen voorafgaand aan het begin van studie behandeling.
- Geen huidige ernstige ongecontroleerde systemische ziekte (bijvoorbeeld klinisch significante cardiovasculaire, pulmonaire of stofwisselingsziekte; wondgenezing stoornissen, zweren, of botbreuken, bekende infectie met HIV, actieve hepatitis B en/of hepatitis C virus) of andere systemische ziekten/symptomen dat naleving van het protocol kan belemmeren, volgens het oordeel van artsen
- Geen grote chirurgische ingreep of ernstig traumatisch letsel binnen 28 dagen voorafgaand aan randomisatie of verwachting dat een grote operatie nodig zal zijn gedurende de studiebehandeling en/of de aanwezigheid van een niet-genezende wond, breuk of een ulcus.
- Geen eerdere ontvangst van studiemedicatie binnen 28 dagen voorafgaand aan randomisatie
- Vruchtbare vrouwen moeten een negatieve serum (of urine) zwangerschapstest hebben 72 uur voorafgaand aan de eerste behandeling
- Patiënten in de vruchtbare/reproductieve leeftijd dienen adequate anticonceptie te gebruiken, zoals gedefinieerd door de onderzoeker, tijdens de studiebehandeling en gedurende ten minste 6 maanden na de laatste studiebehandeling. Een zeer effectieve methode van anticonceptie leidt tot een laag uitvalspercentage (dat wil zeggen minder dan 1% per jaar) mits consequent en correct gebruikt.
- Vrouwen die borstvoeding geven moeten hiermee stoppen voor ontvangst eerste dosis studiemedicatie en tot 6 maanden na ontvangst laatste dosis studiemedicatie.
- Ontbreken van een psychologische, familiaire, sociologische of geografische toestand die mogelijk de naleving van het onderzoeksprotocol en het follow-up schema belemmert; deze voorwaarden moeten voor studieregistratie met de patiënt worden besproken
- Voor registratie/randomisatie moet een getekend informed consent worden afgegeven.
- Weefsel beschikbaarheid voor centrale bevestiging van de histologische diagnose is verplicht. Alle andere TR projecten zijn facultatief voor de patiënt en een apart toestemmingsformulier zal hiervoor beschikbaar worden gesteld.
Exclusiecriteria
- Patiënten die uit de eerstelijns behandeling zijn gehaald door toxiciteit zonder gedocumenteerde ziekteprogressie of die het placebo hebben ontvangen (in de context van een klininische studie) als eerdere behandeling
Studiecoördinator
Prof.dr. T.P. Links, internist-endocrinoloog
Tel.: 050 3613962 (secretariaat Endocrinologie)
Afdeling Endocrinologie
UMCG
Contactpersoon voor de medische oncologie
Dr. S.F. Oosting
Afdeling Medische Oncologie
Tel.: 050 3612821 (secretariaat Oncologie)
Fax: 050 3614862
E-mail: s.f.oosting@umcg.nl