
A Phase 1 Study of the Highly-selective RET Inhibitor, BLU-667, in Patients with Thyroid Cancer, Non-Small Cell Lung Cancer (NSCLC) and Other Advanced Solid Tumors.
| Protocolnummer: | BLU-667-1101 |
| Eudract nummer: | 2016-004390-41 |
| METc nummer: | |
| Onderzoekscode: |
Samenvatting
Dit onderzoek, een fase 1-onderzoek met de selectieve RET-remmer, BLU-667, bij proefpersonen met medullair schildklierkanker (MTC), niet-kleincellige longkanker (NSCLC) en andere uitgezaaide solide tumoren met een RET-alteratie, is opgezet door Blueprint Medicines Corporation en wordt uitgevoerd door artsen in verschillende ziekenhuizen in de US, Azië en Europa. Het doel van dit onderzoek is uitzoeken hoe veilig het nieuwe middel BLU-667 is wanneer het wordt toegediend aan proefpersonen met niet-reseceerbare, uitgezaaide soorten kanker van onder andere de long en schildklier, en het vaststellen van de veilige dosis voor verder onderzoek in fase 2.
Het onderzoeksmiddel BLU-667 wordt getest in verschillende doseringen en in twee delen:
- In het eerste deel wordt uitgezocht wat de meest effectieve dosering van het onderzoeksmiddel is met de minste bijwerkingen. (Deel 1, afgesloten per 03-04-2018)
- In het uitbreidingsgedeelte (deel 2) wordt bevestigd dat de gekozen dosering van het onderzoeksmiddel veilig is en goed wordt verdragen en om te bepalen of het onderzoeksmiddel bij tumoren werkzaam is.
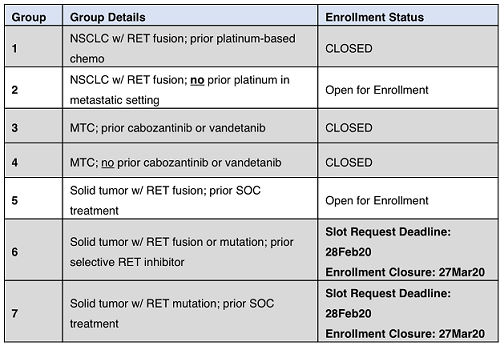
Inclusiecriteria
- Patiënt is ≥ 18 jaar oud
- Diagnose tijdens dosisuitbreiding (deel 2) – alle patiënten in groep 1, 2, 5 en 6 moeten een oncogene RET-verandering/fusie of mutatie (exclusief synonieme en nonsense mutaties) in de tumor hebben, zoals is vastgesteld door plaatselijk testen van de tumor of circulerend tumor-nucleïnezuur in het bloed. Voor alle patiënten in deel 2 van de studie moet tumorweefsel (gearchiveerd of nieuw) beschikbaar zijn voor retrospectief testen van de RET-status. Patienten in groep 6 moeten een nieuw tumorbiopt ondergaan voorafgaand aan deelname.
- Groep 1 – patiënten moeten pathologisch gedocumenteerde, definitief gediagnosticeerde lokaal gevorderde of gemetastaseerde NSCLC met een RET-herschikking hebben die voorheen is behandeld met een MKI.
- Groep 2 – patiënten moeten pathologisch gedocumenteerde, definitief gediagnosticeerde lokaal gevorderde of metastatische NSCLC hebben met een RET-herschikking die niet eerder behandeld werd met een MKI.
- Groep 3 – patiënten moeten pathologisch gedocumenteerde, definitief gediagnosticeerde gevorderde MTC hebben die progressie heeft vertoond binnen 14 maanden voorafgaand aan het screeningsbezoek en welke voorheen is behandeld met een MKI.
- Groep 4 – patiënten moeten pathologisch gedocumenteerde, definitief gediagnosticeerde, gevorderde MTC hebben die binnen 14 maanden voor het screeningsbezoek progressief is en niet eerder behandeld werd met een MKI
- Groep 5 – patiënten moeten een pathologisch gedocumenteerde, definitief gediagnosticeerde, gevorderde solide tumor met een oncogene RET-herschikking/fusie of –mutatie hebben, anders dan NSCLC en METC.
- Groep 6 – patiënten moeten pathologisch gedocumenteerde, definitief gediagnosticeerde, gevorderde solide tumor met een oncogene RET-herschikking/fusie of –mutatie hebben, die eerder werd behandeld met een selectieve TKI die RET remt, zoals LOXO-292.
- De patiënt moet lijden aan een niet-reseceerbare ziekte die gevorderd is na standaardtherapie of die niet adequaat op de standaardtherapie heeft gereageerd, of de patiënt moet intolerant zijn voor de beschikbare standaardtherapieën of deze hebben afgewezen, of er mag geen geaccepteerde standaardtherapie voor zijn ziekte zijn.
- Patiënten in deel 2 van de studie (dosisuitbreiding) moeten meetbare ziekte hebben volgens RECIST v1.1 (of RANO, indien van toepassing voor het tumortype)
- De patiënt stemt toe in het aanleveren van tumorweefsel (opgeslagen, indien beschikbaar, of een nieuwe biopsie) voor bevestiging van RET-status, en is bereid een tumorbiopsie tijdens behandeling te overwegen, indien dit veilig en medisch uitvoerbaar wordt geacht, door de behandelend onderzoeker. Voor deel 2, groep 6, moeten patiënten een nieuwe tumorbiopsie ondergaan om de RET status in het tumorweefsel te bepalen.
- Patiënt heeft een ECOG performance status van 0-1
- De patiënt geeft geïnformeerde instemming om aan het onderzoek deel te nemen.
Exclusiecriteria
- De kanker van de patiënt heeft een bekende primaire drivermutatie anders dan RET. Bijvoorbeeld NSCLC met een mutatie in EGFR, ALK, ROS1 of BRAF waarvoor doelgerichte therapie beschikbaar is; colorectaal carcinoom met een oncogene KRAS-, NRAS- of BRAF-mutatie. Onderzoekers moeten deelname van deze patiënten met co-mutaties met de sponsor bespreken.
- De patiënt heeft één van de volgende binnen 14 dagen voorafgaand aan de eerste dosis van het onderzoeksgeneesmiddel:
- Aantal bloedplaatjes < 75 x 109/l
- Absoluut aantal neutrofielen (ANC) < 1,0 x 109/l
- Hemoglobine < 9.0 g/dl (rode bloedceltransfusie en erytropoëtine kunnen worden gebruikt om ten minste 9,0 g/dl te bereiken, maar moeten ten minste 2 weken voor de eerste dosis van het onderzoeksgeneesmiddel zijn toegediend)
- AST of ALT > 3 x de bovengrens van normaal (ULN) als er geen levermetastasen aanwezig zijn; > 5 x ULN als er levermetastasen aanwezig zijn.
- Totale bilirubine > 1,5 x ULN; > 3 x ULN met directe bilirubine > 1,5 x ULN in aanwezigheid van de ziekte van Gilbert
- Geschatte (Cockroft Gault formule) of gemeten creatinineklaring < 40 ml/min
- De patiënt heeft een QTc > 470 msec. De patiënt heeft een voorgeschiedenis van verlengd QT-syndroom of Torsades de pointes. De patiënt heeft een familiaire voorgeschiedenis van verlengd QT-syndroom.
- De patiënt heeft klinisch significante, ongecontroleerde hart- en vaatziekte, waaronder congestief hartfalen van graad III of IV volgens de classificatie van de NYHA; hartinfarct of instabiele angina in de afgelopen 6 maanden; ongecontroleerde hypertensie of klinisch significante ongecontroleerde aritmieën, waaronder bradyaritmieën die QT-verlenging kunnen veroorzaken (bijv. hartblok van tweede graad van type II of hartblok van derde graad).
- De patiënt heeft metastasen in het centrale zenuwstelsel of een primaire CZS-tumor die geassocieerd wordt met progressieve neurologische symptomen of heeft toenemende doses corticosteroïden nodig om de CZS ziekte te bestrijden. Als een patiënt corticosteroïden nodig heeft voor de behandeling van de CZS ziekte, moet de dosis stabiel zijn geweest gedurende de 2 weken voorafgaand aan C1D1.
- De patiënt onderging anti-kankertherapie (met inbegrip van zowel systemische therapie als radiotherapie) binnen 14 dagen of 5 halfwaardetijden voorafgaand aan de eerste dosis van het onderzoeksgeneesmiddel. BLU-667 kan worden gestart binnen 14 dagen of 5 halfwaardetijden na eerdere behandeling indien de onderzoeker dit veilig en in het belang van de patiënt acht, met voorafgaande toestemming van de sponsor.
- Patiënten met dosisuitbreiding in groepen 1-5 (deel 2): de patiënt werd eerder behandeld met een selectieve RET-remmer zoals LOXO-292.
- De patiënt ontving ondersteunende therapie met neutrofiele groeifactoren binnen 14 dagen voor de eerste dosis van het onderzoeksgeneesmiddel.
- De patiënt heeft behandeling nodig met een verboden medicijn of kruidenmiddel (zie appendix 2 van studieprotocol) dat niet kan worden stop gezet ten minste 2 weken voor aanvang van de toediening van het onderzoeksgeneesmiddel. BLU-667 kan worden gestart binnen 14 dagen of 5 halfwaardetijden na eerdere behandeling indien de onderzoeker dit veilig en in het belang van de patiënt acht, met voorafgaande toestemming van de sponsor.
- De patiënt heeft een majeure chirurgische procedure ondergaan binnen 14 dagen voor de eerste dosis van het onderzoeksgeneesmiddel (procedures zoals het plaatsen van een centrale veneuze katheter, een tumornaaldbiopsie, en het plaatsen van een voedingssonde worden niet beschouwd als majeure chirurgische procedures).
- De patiënt heeft een voorgeschiedenis van een andere primaire maligniteit die in het afgelopen jaar werd gediagnosticeerd of therapie heeft vereist. De volgende eerdere maligniteiten zijn geen exclusiecriterium: volledig gereseceerd basaalcel- en plaveiselcel huidkanker, curatief behandelde lokale prostaatkanker, curatief behandelde lokale schildklierkanker en volledig gereseceerd carcinoma in situ (om het even welke lokatie).
- De patiënt is niet bereid of niet in staat om aan geplande bezoeken, het geneesmiddelentoedieningsplan, laboratoriumtests of andere onderzoeksprocedures en onderzoeksbeperkingen te voldoen.
- Vrouwen die niet bereid zijn, tenzij ze postmenopauzaal of chirurgisch steriel zijn, om zich te onthouden van geslachtsgemeenschap of om zeer effectieve anticonceptie te gebruiken tijdens de periode van toediening van het onderzoeksgeneesmiddel en gedurende ten minste 30 dagen na de laatste dosis van het onderzoeksgeneesmiddel. Mannen die niet bereid zijn, tenzij ze chirurgisch steriel zijn, om zich te onthouden van geslachtsgemeenschap of om zeer effectieve anticonceptie te gebruiken tijdens de periode van toediening onderzoeksgeneesmiddel.
- Zwangere vrouwen, zoals gedocumenteerd door een serum β-hCG zwangerschapstest consistent met zwangerschap, verkregen binnen 7 dagen voorafgaand aan de eerste dosis van het onderzoeksgeneesmiddel. Vrouwen met β-hCG-waarden die binnen het bereik van zwangerschap liggen, maar niet zwanger zijn (fout-positieve uitslagen) kunnen met schriftelijke toestemming van de sponsor worden ingeschreven, nadat zwangerschap werd uitgesloten. Vrouwen die niet-vruchtbaar zijn (langer dan één jaar na de menopauze; bilaterale afbinding van eileiders, bilaterale oophorectomie, hysterectomie) hebben geen serumtest β-hCG nodig.
- Borstvoeding gevende vrouw
- De patiënt heeft eerdere of actuele klinisch significante ziekte, medische toestand, chirurgische voorgeschiedenis, fysieke bevinding, of laboratoriumafwijking die, volgens de onderzoeker, de veiligheid van de patiënt zou kunnen beïnvloeden, de absorptie, de distributie, het metabolisme of de uitscheiding van het onderzoeksgeneesmiddel zou kunnen veranderen, of de beoordeling van onderzoeksresultaten zou kunnen schaden.
Studiecoördinator
Dr. A.J. van der Wekken
Longarts
Tel.: 050 3612942
Voor de Medische Oncologie:
Drs. K.E. Broekman
Afdeling Medische Oncologie
Tel.: 050 3612821 (secretariaat Oncologie)
E-mail: k.e.broekman@umcg.nl